

مرض ساندهوف عند الاطفال
مرض ساندهوف عند الاطفال , داء ساندهوف
اضغط على الرمز  لمشاهدة الصور الايضاحية
لمشاهدة الصور الايضاحية
مرض ساندهوف هو اضطراب وراثي نادر يؤدي إلى تدمير الخلايا العصبية ( الخلايا العصبية ) بشكل تدريجي في الدماغ والحبل الشوكي (الجهاز العصبي المركزي). يتم تصنيف هذه الحالة إلى ثلاثة أنواع رئيسية بناءً على العمر الذي تظهر فيه العلامات والأعراض لأول مرة: الرضع ، والأحداث ، والبالغون.
الشكل الطفولي لمرض ساندهوف هو الشكل الأكثر شيوعًا وشدة ويظهر في مرحلة الطفولة.
أعراض مرض ساندهوف :
عادةً ما يظهر الأطفال المصابون بهذا الاضطراب بشكل طبيعي حتى سن 3 إلى 6 أشهر ، عندما يتباطأ نموهم وتضعف العضلات المستخدمة في الحركة. يفقد الأطفال المتأثرون المهارات الحركية مثل الانقلاب والجلوس والزحف. كما أنهم يطورون رد فعل مفاجئ مبالغ فيه على الضوضاء العالية. مع تقدم المرض ، يعاني الأطفال المصابون بمرض ساندهوف من النوبات وفقدان البصر والسمع والإعاقة الذهنية. من سمات هذا الاضطراب وجود شذوذ في العين يسمى بقعة الكرز الأحمر ، والتي يمكن تحديدها من خلال فحص العين. يعاني بعض الأطفال المصابين أيضًا من ملامح وجه مميزة أو أعضاء متضخمة (تضخم عضوي) أو تشوهات في العظام.
تعد أشكال مرض ساندهوف عند الأحداث والبالغين نادرة جدًا. عادة ما تكون العلامات والأعراض أكثر اعتدالًا من تلك التي تظهر مع الشكل الطفولي ، على الرغم من اختلافها بشكل كبير. يمكن أن يبدأ شكل الأحداث بين سن 2 و 10 سنوات. وتشمل السمات المميزة صعوبات الكلام ، وفقدان الوظيفة الإدراكية (الخرف) ، والنوبات ، وفقدان التنسيق العضلي (الرنح). يتميز مرض Sandhoff للبالغين بمشاكل في الحركة ومشاكل نفسية.
يحدث الشكل الكلاسيكي لمرض ساندهوف عند الرضع.
حيث يبدو أنهم يتمتعون بصحة جيدة عند الولادة ولكنهم يبدأون في إظهار الأعراض في حوالي 3 إلى 6 أشهر من العمر.
قد تشمل العلامات:
- نمو غير طبيعي للعظام.
- حركات محرجة.
- بقع حمراء على مقل العيون.
- تضخم الرأس (تضخم الرأس) أو تضخم الأعضاء الداخلية (تضخم حشوي) ، وخاصة الكبد والطحال.
- ردود فعل مبالغ فيها للضوضاء الصاخبة (استجابة مفاجئة).
- التهابات الجهاز التنفسي المتكررة.
- تطور بطيء في المهارات الحركية ، مثل الزحف والجلوس والتدحرج.
- ضعف العضلات ، صعوبة في السيطرة على العضلات ( ترنح ) أو تشنجات عضلية ( رمع عضلي ).
مع تفاقم مرض ساندهوف ، يعاني الأطفال عادةً من:
- فقدان السمع .
- الإعاقات الذهنية.
- الشلل .
- النوبات.
- فقدان البصر.
- موت مبكر.
نادرًا ما يصاب بعض الأشخاص بمرض ساندهوف في وقت لاحق ، في مرحلة الطفولة أو البلوغ. عادة ما تكون الأعراض متشابهة ولكنها أقل حدة.
الأسباب :
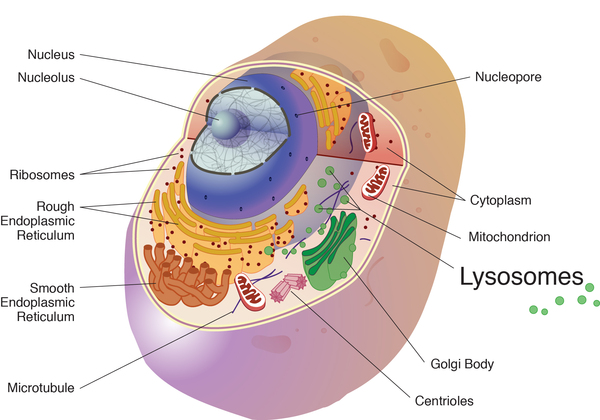 ، وهي هياكل في الخلايا تعمل على تكسير المواد السامة والعمل. كمراكز إعادة التدوير. داخل الجسيمات الحالة ، يقوم هذان الإنزيمان بتفكيك المواد الدهنية والسكريات المعقدة والجزيئات المرتبطة بالسكريات. على وجه الخصوص ، يساعد بيتا هيكسوسامينيداز أ على تكسير مادة دهنية تسمى GM2 ganglioside.
، وهي هياكل في الخلايا تعمل على تكسير المواد السامة والعمل. كمراكز إعادة التدوير. داخل الجسيمات الحالة ، يقوم هذان الإنزيمان بتفكيك المواد الدهنية والسكريات المعقدة والجزيئات المرتبطة بالسكريات. على وجه الخصوص ، يساعد بيتا هيكسوسامينيداز أ على تكسير مادة دهنية تسمى GM2 ganglioside.تعمل المتغيرات في جين HEXB على تعطيل نشاط بيتا هيكسوسامينيداز أ وبيتا هيكسوسامينيداز ب ، مما يمنع هذه الإنزيمات من تكسير جزيئات معينة ، بما في ذلك GM2 ganglioside. نتيجة لذلك ، يمكن أن تتراكم هذه المركبات إلى مستويات سامة ، خاصة في الخلايا العصبية في الدماغ والحبل الشوكي. على وجه الخصوص ، يؤدي تراكم GM2 ganglioside إلى التدمير التدريجي لهذه الخلايا العصبية ، مما يتسبب في العديد من علامات وأعراض مرض Sandhoff. تحدد شدة النقص (النقص) في إنزيمات بيتا هيكسوسامينيداز أ وب عادة العمر الذي تحدث فيه السمات وشكل مرض ساندهوف الذي يتطور.
نظرًا لأن مرض Sandhoff يضعف وظيفة الإنزيمات الليزوزومية وينطوي على تراكم GM2 ganglioside ، يُشار إلى هذه الحالة أحيانًا باسم اضطراب التخزين الليزوزومي أو داء الجراثيم GM2.
مرض ساندهوف هو واحد من ثلاثة أمراض ناتجة عن تراكم GM2 ganglioside. يُطلق على المرضين الآخرين اسم مرض تاي ساكس وداء الغنغليوزيدات GM2 ، وهو متغير AB ، وينجم عن متغيرات في جينات أخرى.
يتم توريث هذه الحالة في نمط وراثي جسمي متنحي ، مما يعني أن كلا نسختي الجين في كل خلية لهما متغيرات. يحمل كل من والدي الفرد المصاب بحالة جسمية متنحية نسخة واحدة من الجين المتغير ، لكنهم عادة لا يظهرون علامات وأعراض الحالة.
، مما يعني أن كلا نسختي الجين في كل خلية لهما متغيرات. يحمل كل من والدي الفرد المصاب بحالة جسمية متنحية نسخة واحدة من الجين المتغير ، لكنهم عادة لا يظهرون علامات وأعراض الحالة.